





Histone Dimethylation Analysis Service
Based on a high-resolution liquid chromatography-tandem mass spectrometry (LC-MS/MS) platform, the histone dimethylation analysis service launched by MtoZ Biolabs enables systematic detection and quantitative analysis of dimethylation modifications on lysine or arginine residues of histones. This service can accurately identify dimethylation modifications at different sites and analyze their abundance changes under different conditions. By integrating bioinformatics methods, it further provides data on modification site distribution, differential comparison, and functional annotation, offering reliable support for researchers to explore the role of dimethylation in epigenetic regulation and cellular function studies.
Overview
Histone dimethylation (me2) refers to a type of post-translational modification in which two methyl groups are attached to lysine or arginine residues of histones, playing a critical role in maintaining chromatin structure and regulating gene transcription. Dimethylation at different sites is often closely associated with gene activation or repression and has important functions in biological processes such as cell cycle regulation, differentiation and development, metabolic adaptation, and stress response. Histone dimethylation analysis enables systematic detection and quantitative analysis of dimethylation modifications and is widely applied in epigenetics, cellular function research, and potential biomarker exploration, providing reliable data support for a deeper understanding of its biological significance.
Analysis Workflow
1. Histone Extraction and Digestion
Histones are isolated from cell or tissue samples and enzymatically digested under optimized conditions to generate detectable peptides.
2. Modified Peptide Enrichment
Specific enrichment methods are used to capture dimethylated peptides, improving detection rate and coverage.
3. LC-MS/MS Analysis
High-resolution mass spectrometry is applied for precise identification and quantitative analysis of dimethylation modification sites.
4. Data Analysis
Bioinformatics methods are used to provide results on modification distribution, abundance changes, and functional annotation.
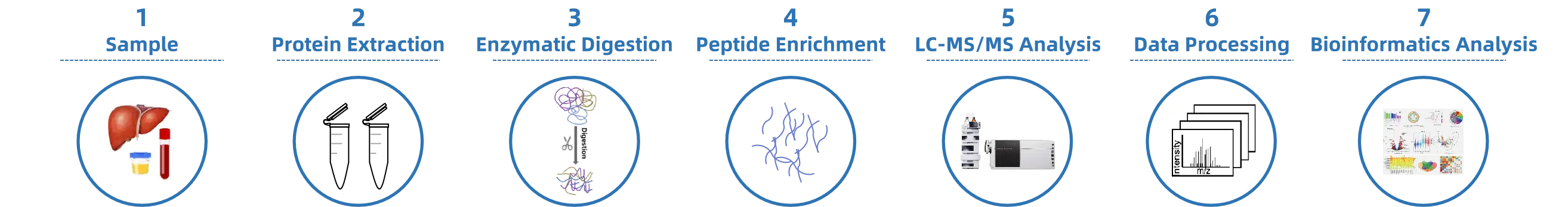
Figure 1. The Workflow of Histone Dimethylation Analysis.
Sample Submission Suggestions
1. Sample Type and Quantity
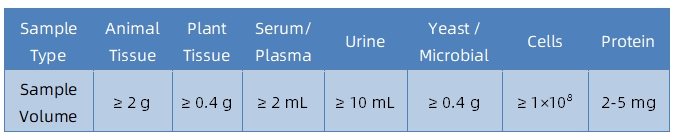
Note: Plasma should be collected using EDTA as an anticoagulant. Standard tissue or cell lysis buffers can be used during protein extraction.
2. Sample Transportation
Avoid repeated freeze-thaw cycles. Samples are recommended to be stored at -80°C and transported on dry ice to ensure low-temperature conditions throughout the process and prevent modification loss.
Service Advantages
1. High-Resolution Detection
Relying on an advanced LC-MS/MS platform to ensure accurate identification of low-abundance dimethylation modifications.
2. Efficient Enrichment Strategy
Using optimized peptide enrichment methods to significantly improve detection sensitivity and coverage.
3. Strict Quality Control
A standardized quality control system is implemented throughout the entire process to ensure data stability and reproducibility.
4. Flexible Experimental Design
Experimental workflows can be customized according to different sample types and research objectives to meet diverse needs.
Applications
1. Epigenetic Regulation Research
Histone dimethylation analysis service can be used to elucidate the role of dimethylation modifications in gene silencing or activation mechanisms.
2. Environmental Response Research
By analyzing modification level changes, reveal the regulatory role of dimethylation under nutrient shifts, stress, and external stimuli.
3. Cell Cycle Research
Histone dimethylation analysis service can be applied to study the dynamic regulation of dimethylation in cell cycle progression.
4. Potential Biomarker Discovery
Identify representative dimethylation modification features under specific conditions to provide a basis for biomarker development.
FAQ
Q1: What Is the Difference between Dimethylation, Monomethylation, and Trimethylation?
A1: Dimethylation is a modification in which two methyl groups are attached to lysine or arginine residues of histones, usually associated with gene silencing or specific activation states. In contrast, monomethylation and trimethylation differ in regulatory functions and site specificity, making it important to distinguish them in analysis.
Q2: Which Histone Dimethylation Sites Can This Service Analyze?
A2: It can cover dimethylation sites on multiple histone subtypes (such as H3 and H4), including common ones like H3K9me2 and H3K27me2, and can be expanded to other sites depending on research needs.
Q3: Does This Service Support Multi-Omics Integrated Analysis?
A3: Yes. Dimethylation analysis can be combined with transcriptomics, metabolomics, or other epigenetic modifications to construct a more comprehensive regulatory network.


