





Protein Methylation Analysis Service
Based on advanced high-resolution mass spectrometry platforms and specific enrichment strategies, MtoZ Biolabs has launched the protein methylation analysis service which enables comprehensive identification and quantification of methylation sites in protein samples. This service can analyze the site distribution, modification levels, and dynamic changes of methylation, and perform pathway and functional association analysis through bioinformatics methods. The resulting data can help researchers gain a deeper understanding of the functional characteristics of proteins within regulatory networks, providing a solid reference for subsequent mechanistic exploration and experimental design.
Overview
Protein methylation refers to the process in which methyl groups are introduced into lysine or arginine residues of proteins catalyzed by methyltransferases. This modification can regulate protein conformation, interactions, and stability, thereby affecting multiple biological processes such as gene expression, signal transduction, and the cell cycle. The principle of protein methylation analysis mainly relies on mass spectrometry technology combined with specific enrichment or immunological methods to achieve precise identification and quantification of methylation sites, revealing their role in cellular functional regulation. This method is widely applied in signaling pathway research, epigenetic studies, and the analysis of drug action mechanisms, providing data support for a deeper understanding of life processes and the development of novel drugs.
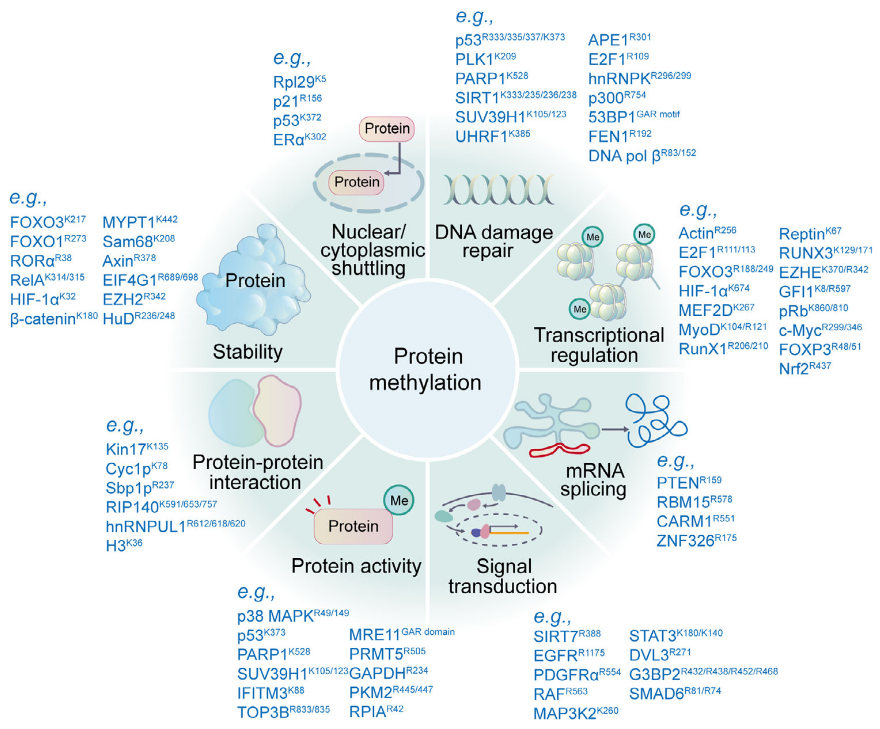
Zhong, Q. et al. MedComm (2020). 2023.
Figure 1. Functions of Protein Methylation.
Services at MtoZ Biolabs
1. Target Protein Methylation Analysis
MtoZ Biolabs can perform methylation detection on specific target proteins, including lysine or arginine site modifications, and analyze modification types and site distribution according to experimental needs. Powered by a high-resolution LC-MS/MS platform, we can accurately compare site-specific methylation changes under different treatment conditions, providing researchers with clear regulatory insights into the target protein.
2. Methylation Proteomics Analysis
MtoZ Biolabs utilizes methylated-peptide enrichment strategies (such as anti-methylation antibody enrichment and HILIC) combined with large-scale mass spectrometry to achieve system-level profiling of methylation modifications across the entire proteome. This analysis enables simultaneous identification and quantification of multiple types of methylation events, revealing their distribution patterns within protein regulatory networks and providing comprehensive data support for downstream functional studies and mechanism exploration.
Analysis Workflow
1. Sample Preparation
Protein extraction and quantification of samples such as cells, tissues or body fluids to ensure uniform quality.
2. Protein Digestion
Proteins are digested into peptides to facilitate the selective detection of methylation modifications.
3. Methylated Peptide Enrichment
Specific antibodies or affinity chromatography methods are used to enrich methylated peptides, improving detection sensitivity and specificity.
4. Mass Spectrometry Detection
Based on high-resolution LC-MS/MS platforms, enriched peptides are subjected to high-precision identification and quantitative analysis.
5. Data Analysis
Database searches and bioinformatics tools are applied to annotate methylation sites and provide output on distribution characteristics, modification levels, and potential functional information.
Sample Submission Suggestions
1. Sample Type and Quantity
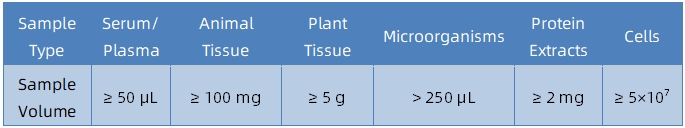
2. Sample Storage
Samples should be stored under low-temperature conditions (such as -80°C freezing) to avoid repeated freeze-thaw cycles and to prevent protein degradation and loss of methylation modifications.
3. Sample Transportation
During transportation, dry ice or cold chain conditions should be used to ensure that samples maintain integrity and modification stability before arriving at the testing platform.
Service Advantages
1. High Sensitivity
Utilizing advanced LC-MS/MS platforms, the service enables accurate detection of low-abundance methylation sites in complex biological samples.
2. Specific Enrichment
Multiple enrichment approaches, such as antibody-based or affinity methods, ensure effective isolation of methylated peptides and improve data accuracy.
3. Professional Expertise
A team with extensive experience in protein modification and mass spectrometry ensures standardized operations and reliable data interpretation.
4. Customized Solutions
Flexible analytical strategies are provided to meet diverse research needs, ensuring adaptability across different experimental objectives.
Applications
1. Signal Pathway Research
Protein methylation analysis service helps to study how methylation regulates signaling pathways and protein interactions.
2. Epigenetic Studies
The service supports exploration of protein-level methylation involved in gene expression regulation.
3. Cell Cycle Control
Analysis reveals the role of methylation in cell cycle progression and fundamental cellular functions.
4. Biomarker Discovery
The protein methylation analysis service provides data for identifying potential markers in basic biological research.
FAQ
Q1: Will Improper Sample Storage Affect the Results?
A1: Yes. If samples undergo repeated freeze-thaw cycles or are stored under unsuitable conditions, protein degradation or loss of modifications may occur, thereby affecting the accuracy of detection.
Q2: Is Methylation Modification Easily Lost during Mass Spectrometry Analysis?
A2: During sample preparation and analysis, some modifications may be affected by digestion efficiency or ionization conditions. By using optimized digestion protocols and detection workflows, we maximize the retention of modification information.
Q3: Will Protein Methylation Interfere with the Detection of Other Post-Translational Modifications?
A3: In complex samples, methylation may coexist with modifications such as acetylation and phosphorylation. Mass spectrometry can distinguish these, but requires high-resolution detection and robust data analysis workflows.


